Central Research Approach: exploiting chromatin-remodeling complexes to study vascular development and maintenance
For the lab’s first decade, we focused on the role of two key ATP-dependent chromatin-remodeling complexes in vascular development: SWI/SNF and NuRD. We first reported that these complexes transcriptionally and antagonistically regulate yolk sac vascular Wnt signaling at midgestation (Griffin and Curtis et al, 2011; Curtis and Griffin, 2012). Further analysis of SWI/SNF mutants at later stages of development revealed that they had defective venous specification due to misregulated expression of the venous transcription factor COUP-TFII (Davis et al, 2013) and defective capillary integrity due to misregulated expression of the serum response factor cofactors MRTFA/B (Menendez and Ong et al, 2017). These studies are important because they were among the first to reveal the contribution of chromatin-remodeling complexes to vascular development, and they established a model for how we could glean interesting genetic and molecular information from vascular phenotypes in chromatin-remodeling complex mutants. Notably, our approach has been tractable and productive because we can frequently attribute the specific phenotypes generated in our mutants to a single misregulated gene or signaling pathway. Therefore, this unbiased approach is revealing genes for which tight transcriptional regulation or timing is essential during vascular development and maintenance.
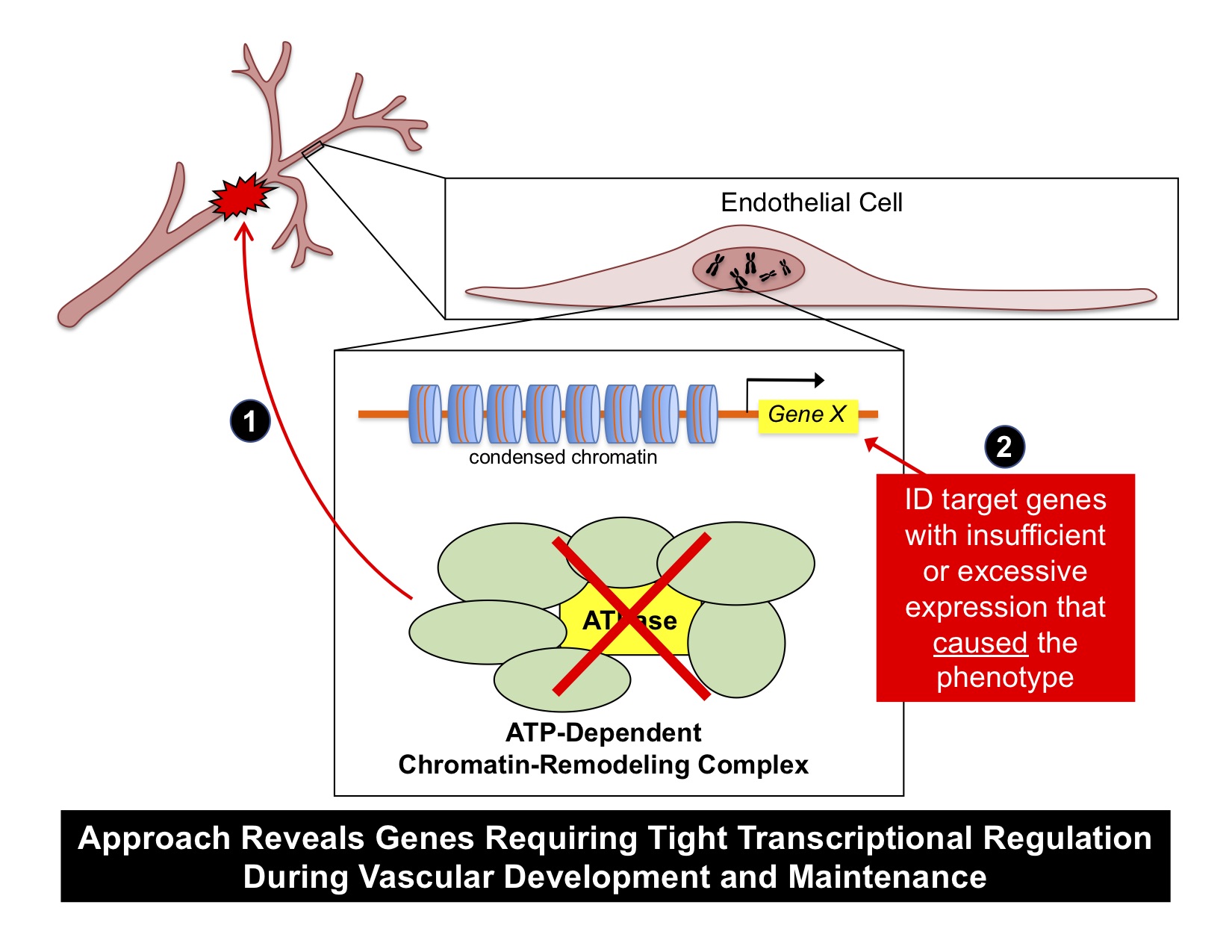
We exploit ATP-dependent chromatin-remodeling complexes to identify critical genes for vascular biology. We genetically delete the central ATPase of these complexes (which is critical for their function) in murine endothelial cells in vivo. We then look for a vascular phenotype that reproducibly correlates with the mutation (1). Finally, we use candidate or unbiased approaches to identify the gene(s) that were misregulated by the mutation and that caused the vascular phenotype (2). This tractable approach combines strengths of forward and reverse genetics and reveals genes requiring tight transcriptional regulation during vascular development and maintenance.
See these tabs for descriptions of specific research areas in the lab–some of which have emerged from the approach described above:
Plasmin-mediated vascular fragility
RIPK3 and vascular biology
Vascular regression